
Redes Neurais e sua Aplicação na Previsão de Propriedades de Materiais Fotovoltaicos
Dennys Felipe Oliveira Santicioli Rizzon*
Com a crescente preocupação com os problemas ambientais e a necessidade de reduzir as emissões de carbono, a busca por fontes de energia limpa e renovável tornou-se uma prioridade global [2]. No entanto, o desenvolvimento de novas tecnologias energéticas depende da descoberta de materiais avançados, capazes de converter e armazenar energia de forma eficiente [1]. Tradicionalmente, a descoberta de novos materiais dependia de experimentos laboratoriais e cálculos teóricos, como os métodos baseados na Teoria do Funcional da Densidade (DFT). Embora precisos, esses métodos podem ser demorados e exigem elevado poder computacional [3]. No entanto, o avanço do aprendizado de máquina (ML), tem revolucionado a área de materiais, oferecendo uma nova abordagem para acelerar a descoberta e o projeto de materiais energéticos.
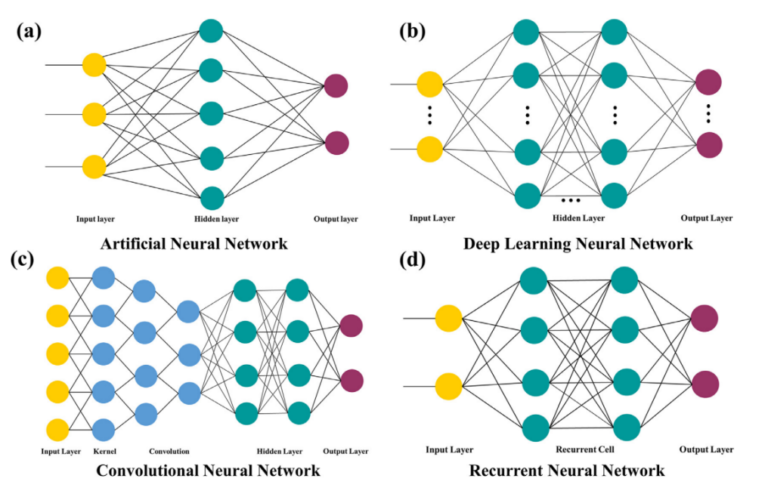
2 Redes Neurais
As redes neurais artificiais são modelos computacionais inspirados no funcionamento do cérebro humano, são modelos computacionais projetados para reconhecer
padrões e estabelecer relações complexas entre dados.
Uma rede neural artificial típica, ilustrada na figura 1.a, é composta por três camadas principais: a camada de entrada, a camada oculta (ou camadas ocultas, no caso de redes profundas) e a camada de saída. A camada de entrada representa a variável independente, que pode assumir diferentes valores e atributos conforme o caso. A camada oculta realiza uma transformação não linear, aplicando pesos e transmitindo os dados processados à saída via função de ativação, cuja estrutura varia conforme a função da rede. Já a camada de saída representa a variável dependente, que também serve como o alvo da rede neural artificial.
Modelos mais sofisticados, como as redes neurais profundas apresentadas na figura 1.b, possuem múltiplas camadas ocultas, permitindo a modelagem de sistemas extremamente complexos. Além disso, variações como as redes neurais convolucionais e recorrentes têm se mostrado eficazes em tarefas como a previsão de propriedades de materiais e na triagem de compostos promissores
3 Desenvolvimento de Materiais Fotovoltaicos
A energia solar é uma das fontes renováveis mais promissoras para a transição energética global. Para tornar sua adoção mais eficiente, é essencial desenvolver materiais fotovoltaicos que maximizem a conversão de luz solar em eletricidade. Os algoritmos de ML, como as redes neurais, têm sido empregados para prever propriedades essenciais e identificar novas composições que podem revolucionar a área fotovoltaica.
Essa abordagem permite não só a previsão de características importantes, como estabilidade e eficiência de conversão, mas também a identificação de estruturas cristalinas mais eficientes. A integração entre as técnicas de aprendizado de máquina e os cálculos teóricos, como o DFT, possibilita uma triagem rápida e precisa dos materiais. Por exemplo, métodos de classificação, aplicados para prever a estabilidade de estruturas como as perovskitas [5], já demonstraram altas taxas de acerto, direcionando os pesquisadores para os compostos com maior potencial para aplicação em células solares. Essa estratégia de validação mútua entre teoria e experimentação acelera o processo de descoberta, contribuindo para a evolução dos dispositivos fotovoltaicos.
4 Perspectivas Futuras:
Apesar dos avanços significativos, alguns desafios ainda precisam ser superados para que essas tecnologias alcancem seu pleno potencial. Um dos maiores obstáculos é a limitação na disponibilidade de dados, tanto experimentais quanto teóricos, uma vez que a maioria dos modelos de aprendizado de máquina depende de grandes volumes de informação para fazer previsões precisas.
Para superar esse problema, novas abordagens, como a mineração de texto e o reconhecimento de imagens, têm sido exploradas para extrair dados de artigos científicos e experimentos computacionais. Além disso, combinar o aprendizado de máquina com a teoria funcional da densidade pode melhorar a precisão na previsão de propriedades dos materiais, reduzindo significativamente o tempo necessário para identificar novos candidatos.
Com o avanço das técnicas de aprendizado profundo e a ampliação das bases de dados disponíveis, a descoberta de materiais inovadores pode se tornar cada vez mais eficiente e acessível.
[1] Peng Gao et al. “The Role of Cation Vacancies in Electrode Materials for Enhanced Electrochemical Energy Storage: Synthesis, Advanced Characterization, and Fundamentals”. Em: Advanced Energy Materials 10 (2020). doi: 10 . 1002 / aenm.201903780.
[2] Donghan Jin et al. “Energy and AI”. Em: Energy and AI 1 (2020). doi: 10.1016/j.egyai.2020. 100002.
[3] Yongqiang Kang, Lejing Li e Baohua Li. “Recent progress on discovery and properties prediction of energy materials: Simple machine learning meets complex quantum chemistry”. Em: Journal of Energy Chemistry 54 (2021). doi: 10.1016/j.jechem.2020.05.044.
[4] Yun Liu et al. “Machine learning for advanced energy materials”. Em: Energy and AI 3 (2021). doi: https://doi.org/10.1016/j.egyai. 2021.100049.
[5] Shuaihua Lu et al. “Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning”. Em: Nature Communications 9 (2018). doi: 10.1038/s41467- 018- 05761-w.
Autor:
Dennys Felipe Oliveira Santicioli Rizzon , aluno de mestrado bolsista NAPI EZC. Desenvolve o projeto sob a orientação da Prof Dr Jose Eduardo Padilha de Sousa, docente da Universidade Federal do Paraná
Essa pesquisa
contribui para as seguintes ODS:




